Books


Projects

 SAnDReS: Statistical Analysis of Docking Results and Scoring functions
SAnDReS: Statistical Analysis of Docking Results and Scoring functions


 Taba: A Tool to Analyze the Binding Affinity
Taba: A Tool to Analyze the Binding Affinity
Citation

Editorships

Frontiers Section Editor (Bioinformatics and Biophysics) for the Current Drug Targets ISSN: 1873-5592
![]()

Section Editor (Bioinformatics in Drug Design and Discovery) for the Current Medicinal Chemistry ISSN: 1875-533X
![]()

Member of the Editorial Board of the Journal of Molecular Structure ISSN: 0022-2860
![]()

Member of the Editorial Board of Molecular Diversity ISSN: 1381-1991 (Print) 1573-501X (Online)
![]()

Associate Editor for Exploration of Drug Science
![]()

Reviewer Editor for Frontiers in Chemistry ISSN: 2296-2646
![]()

Member of the Editorial Board for the Organic and Medicinal Chemistry International Journal ISSN: 2474-7610
As highlighted in a seminal paper by Kitano, 2002: "Computational systems biology addresses questions fundamental to our understanding of life, yet progress here will lead to practical innovations in medicine, drug discovery, and engineering." With this view, we aim to merge the holistic approach of systems biology with machine-learning methods to contribute to drug discovery projects (Bitencourt-Ferreira et al., 2023). Voit says biological systems generally contain several units, but they present an additional challenge to any study because the processes that govern them are not linear (Voit, 2018). For instance, the use of Biochemical Systems Theory (Voit & Savageau, 1987) allows us to model complex biological systems to predict oscillatory patterns (e.g., cyclin-dependent kinase 2) (Goldbeter, 2013) (Figure below) and other behaviors.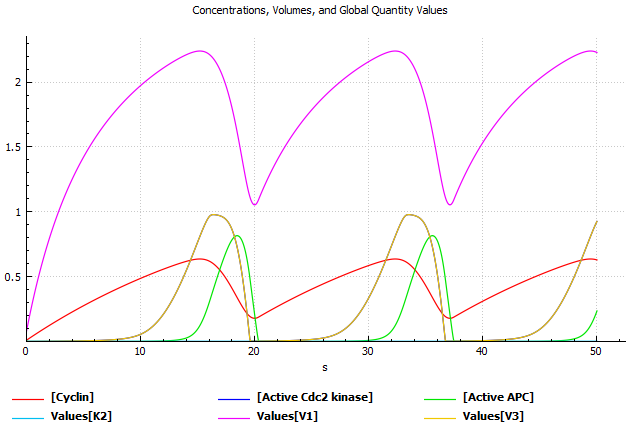
Model for oscillations of Cdc kinase activity in embryonic cell cycles based on Michaelis-Menten phosphorylation-dephosphorylation kinetics (Goldbeter, 2013) (SBML model available here). Square brackets indicate concentrations. Vi denotes the effective maximum rate for each of the enzymes. APC means anaphase-promoting complex. COPASI (Mendes et al., 2009) generated the above plot.
We have been working on computational models to predict enzyme inhibition and protein-ligand interactions (Bitencourt-Ferreira & de Azevedo, 2019a; 2019b; 2019c; da Silva et al., 2020). We can use these computational models to predict the binding affinity of a potential inhibitor for an enzyme; such knowledge speeds up drug discovery and decreases the cost of new drugs (de Ávila et al., 2017; Pintro & Azevedo, 2017).
Furthermore, the availability of computational models to predict binding affinity based on the atomic coordinates of protein-ligand complexes adds flexibility to drug discovery (Xavier et al., 2016; Heck et al., 2017). It allows us to computationally test different scenarios where a potential new drug may interact with a protein target (Bitencourt-Ferreira & de Azevedo, 2021). We developed the programs SAnDReS (Xavier et al., 2016; Bitencourt-Ferreira et al., 2021) and Taba (da Silva et al., 2020, Bitencourt-Ferreira et al., 2021) to create machine-learning models targeted to the protein system of interest. We have successfully employed SAnDReS to study coagulation factor Xa (EC 3.4.21.6) (Xavier et al., 2016), cyclin-dependent kinases (EC 2.7.11.22) (de Ávila et al., 2017; Levin et al., 2018), HIV-1 protease (EC 3.4.23.16) (Pintro & de Azevedo, 2017), estrogen receptor (Amaral et al., 2018), cannabinoid receptor 1 (Russo & de Azevedo, 2019; Russo & de Azevedo, 2020), and 3-dehydroquinate dehydratase (EC 4.2.1.10) (de Ávila & de Azevedo, 2018). Also, we used SAnDReS (Xavier et al., 2016) to develop a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes (Bitencourt-Ferreira & de Azevedo Jr., 2018).