Books


Projects

 SAnDReS: Statistical Analysis of Docking Results and Scoring functions
SAnDReS: Statistical Analysis of Docking Results and Scoring functions


 Taba: A Tool to Analyze the Binding Affinity
Taba: A Tool to Analyze the Binding Affinity
Citation

Editorships

Frontiers Section Editor (Bioinformatics and Biophysics) for the Current Drug Targets ISSN: 1873-5592
![]()

Section Editor (Bioinformatics in Drug Design and Discovery) for the Current Medicinal Chemistry ISSN: 1875-533X
![]()

Member of the Editorial Board of the Journal of Molecular Structure ISSN: 0022-2860
![]()

Member of the Editorial Board of Molecular Diversity ISSN: 1381-1991 (Print) 1573-501X (Online)
![]()

Associate Editor for Exploration of Drug Science
![]()

Reviewer Editor for Frontiers in Chemistry ISSN: 2296-2646
![]()

Member of the Editorial Board for the Organic and Medicinal Chemistry International Journal ISSN: 2474-7610
Highlights
Taba (Tool to Analyze the Binding Affinity) generates scoring functions to predict binding affinity based on the atomic coordinates of a protein-ligand complex. It employs a polynomial equation where the terms are mass-spring potentials. Taba calculates average interatomic distances and takes them as equilibrium distances for a mass-spring potential. The mass-spring potential equation and the equilibrium constants for each pair of atoms compose the Taba Force Field (TFF). We are currently updating Taba and expect to release a new version of Taba by July/2024. It is indicated below a flowchart highlighting the main steps of Taba. Please cite the following reference (da Silva et al., 2020) if you use the Taba program.
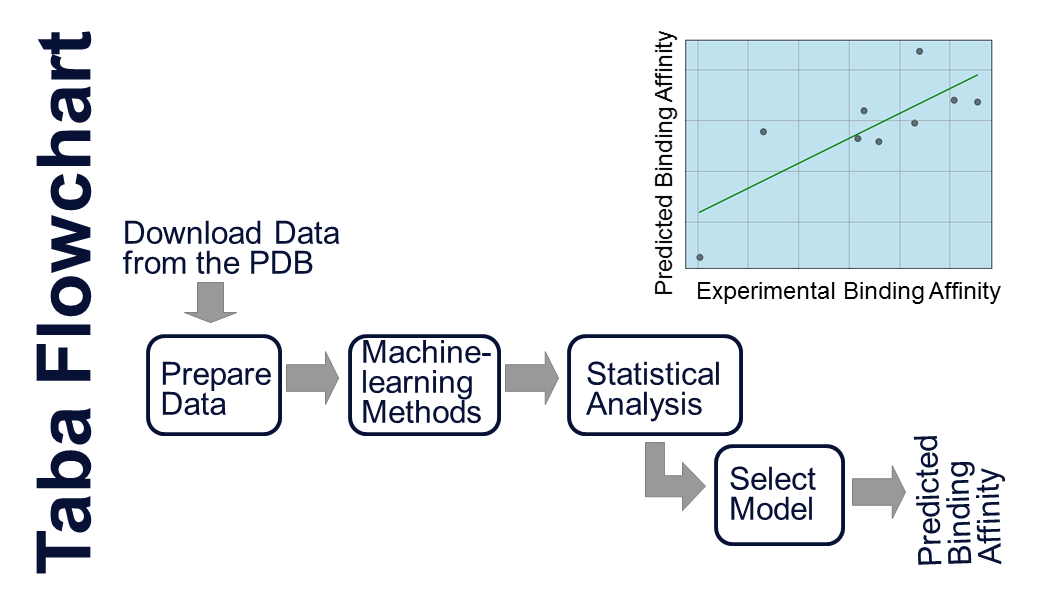
This flowchart shows the main steps used to generate targeted-scoring functions with Taba (da Silva et al., 2020).
TFF outperforms classical and machine learning scoring functions such as KDEEP (Jiménez et al., 2018) and CSM-lig (Pires & Ascher, 2016)(plot below).
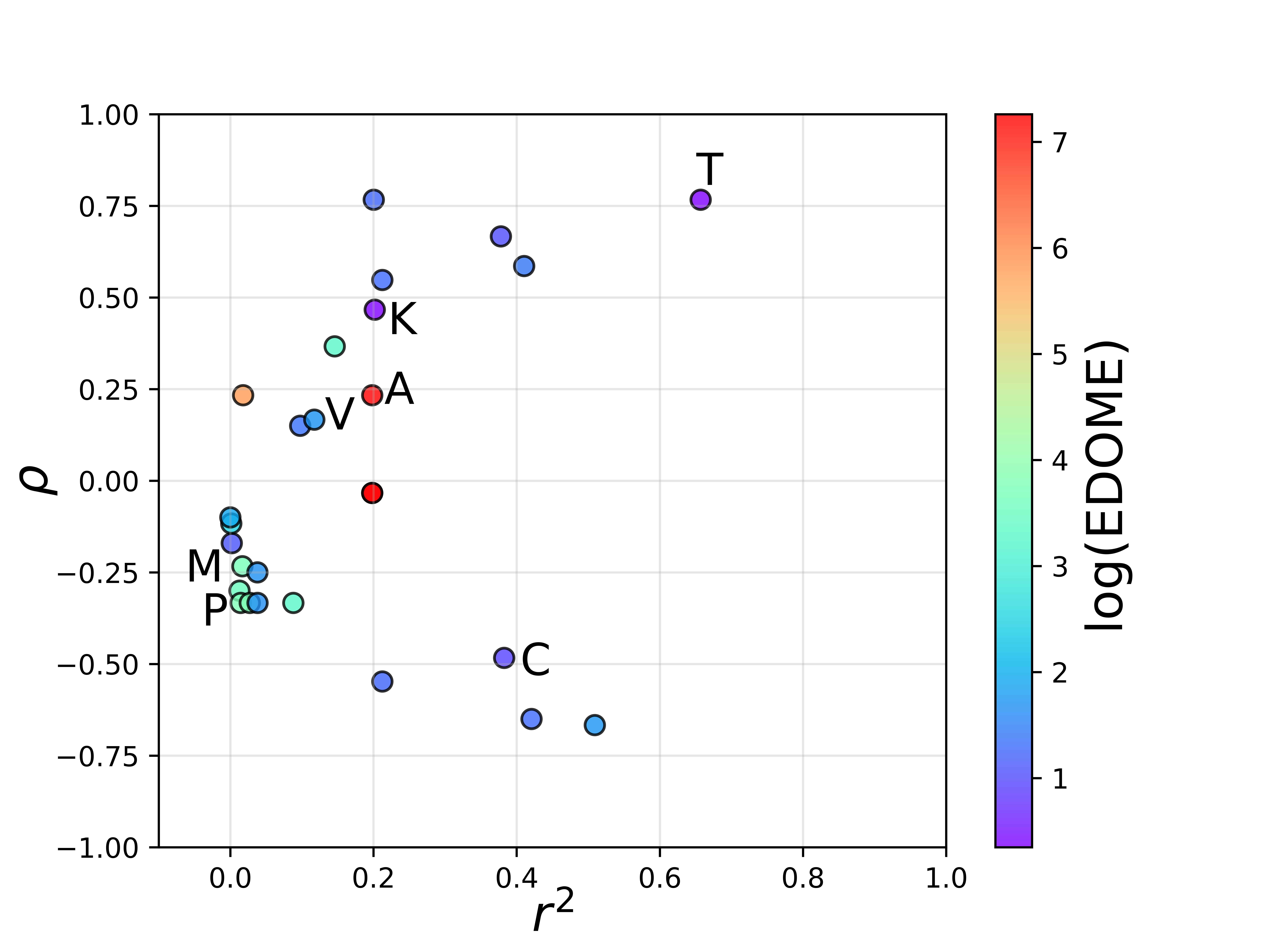
Predictive performance with DOME (Walsh et al., 2021) using r2, rho, and log (EDOME). We employed EDOME to generate this plot. Statistical analysis of scoring functions includes previously published data (da Silva et al., 2020) to create this plot, except for KDEEP (Jiménez et al., 2018) and CSM-lig (Pires & Ascher, 2016), which utilized the atomic coordinates for the structures in the test set to predict the affinity (pKi). The plot highlights the performance of seven scoring functions: T (Taba), K (KDEEP ), C (CSM-lig), A (AutoDock4) (Morris et al. 2009), V (AutoDock Vina) (Eberhardt et al., 2021), M (MolDock Score), and P (Plants Score) (Thomsen & Christensen, 2006).