Books


Projects

 SAnDReS: Statistical Analysis of Docking Results and Scoring functions
SAnDReS: Statistical Analysis of Docking Results and Scoring functions


 Taba: A Tool to Analyze the Binding Affinity
Taba: A Tool to Analyze the Binding Affinity
Citation

Editorships

Frontiers Section Editor (Bioinformatics and Biophysics) for the Current Drug Targets ISSN: 1873-5592
![]()

Section Editor (Bioinformatics in Drug Design and Discovery) for the Current Medicinal Chemistry ISSN: 1875-533X
![]()

Member of the Editorial Board of the Journal of Molecular Structure ISSN: 0022-2860
![]()

Member of the Editorial Board of Molecular Diversity ISSN: 1381-1991 (Print) 1573-501X (Online)
![]()

Associate Editor for Exploration of Drug Science
![]()

Reviewer Editor for Frontiers in Chemistry ISSN: 2296-2646
![]()

Member of the Editorial Board for the Organic and Medicinal Chemistry International Journal ISSN: 2474-7610
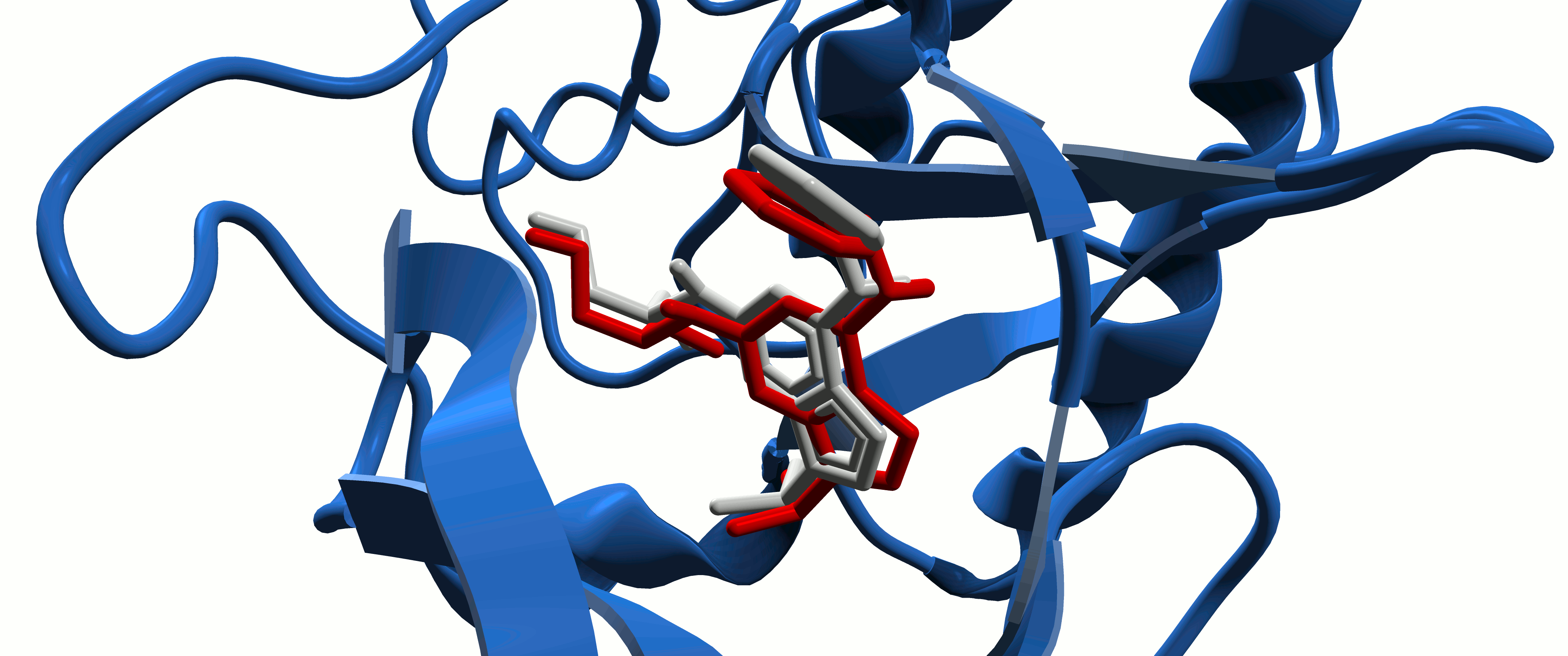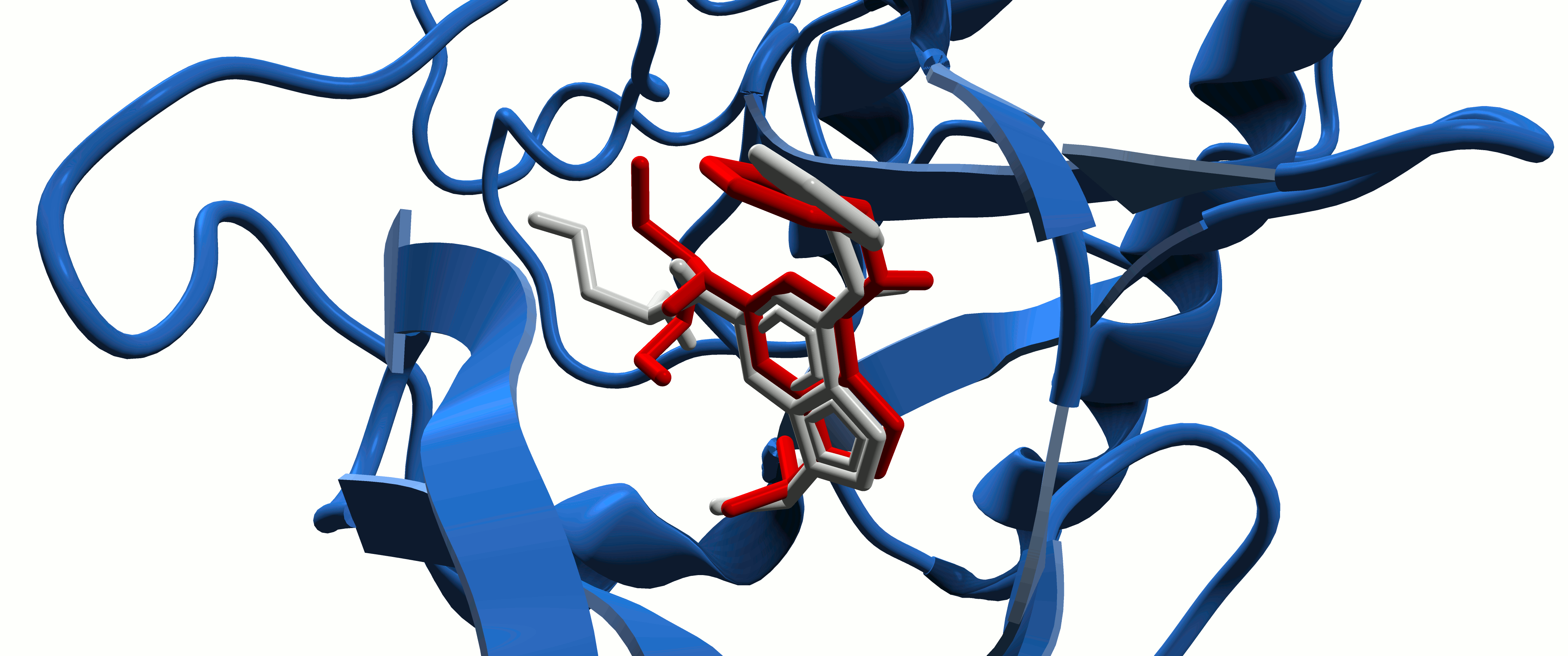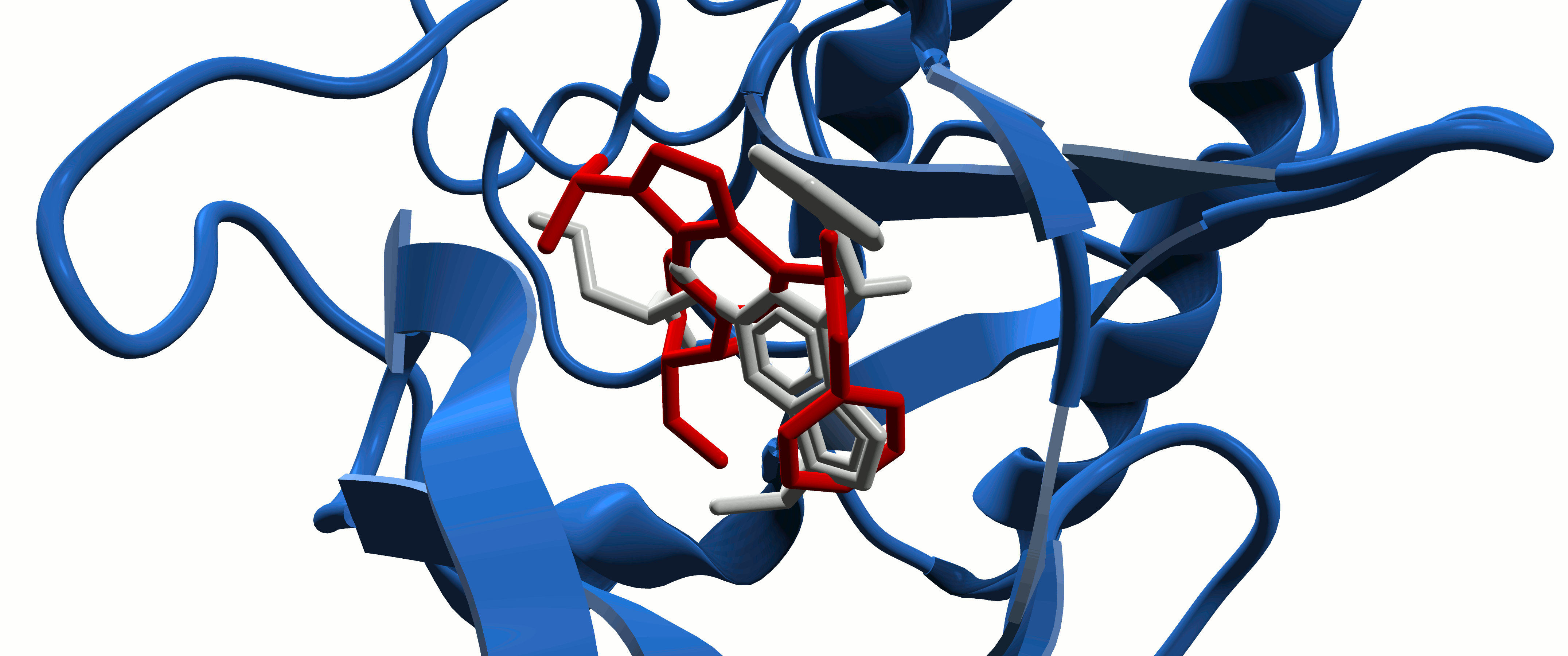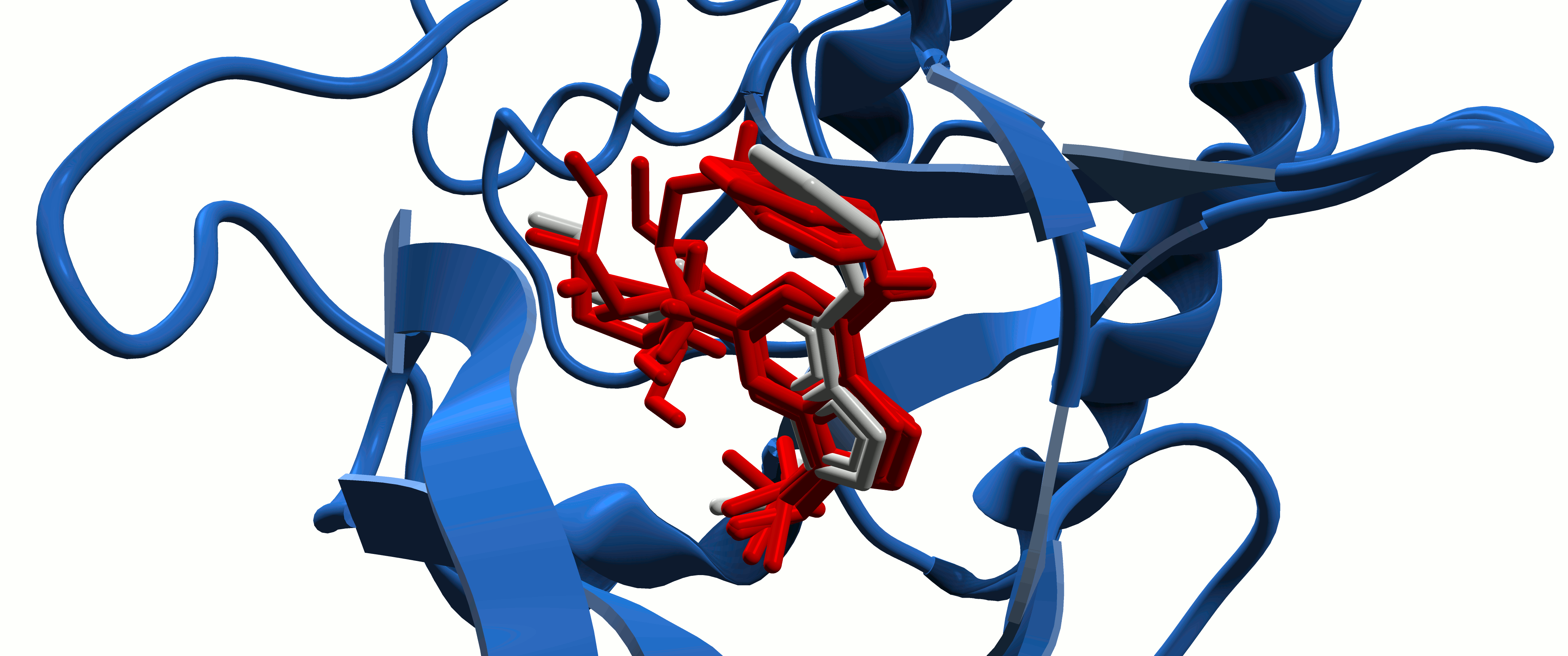
In the study of intermolecular interactions involving protein and ligands, we expect to gain further insights into the structural basis for the specificity of small-molecule ligands against a specific protein target (de Azevedo, 2008). Analysis of protein-ligand interaction is a central problem in drug design. Knowledge of the key features responsible for the specificity of a ligand for a protein allows us to determine which physical-chemical parameters could improve the protein-ligand interaction (de Azevedo & Dias, 2008a). Furthermore, the development of a computational model to predict the binding affinity based on the atomic coordinates of a protein-ligand complex opens the possibility to apply virtual screening approaches to search small-molecule databases to identify a drug candidate (de Azevedo & Dias, 2008b, de Azevedo, 2010a; Bitencourt-Ferreira & de Azevedo, 2019a; da Silva et al., 2020 ). To study protein-ligand interactions, we make use of protein crystallography (Canduri & de Azevedo, 2008; Veit-Acosta & de Azevedo, 2021), nuclear magnetic resonance spectroscopy (Fadel et al., 2005), molecular docking (de Azevedo, 2010b), and molecular dynamics (de Azevedo, 2011; Bitencourt-Ferreira & de Azevedo, 2019b).

 d
d