Books


Projects

 SAnDReS: Statistical Analysis of Docking Results and Scoring functions
SAnDReS: Statistical Analysis of Docking Results and Scoring functions


 Taba: A Tool to Analyze the Binding Affinity
Taba: A Tool to Analyze the Binding Affinity
Citation

Editorships

Frontiers Section Editor (Bioinformatics and Biophysics) for the Current Drug Targets ISSN: 1873-5592
![]()

Section Editor (Bioinformatics in Drug Design and Discovery) for the Current Medicinal Chemistry ISSN: 1875-533X
![]()

Member of the Editorial Board of the Journal of Molecular Structure ISSN: 0022-2860
![]()

Member of the Editorial Board of Molecular Diversity ISSN: 1381-1991 (Print) 1573-501X (Online)
![]()

Associate Editor for Exploration of Drug Science
![]()

Reviewer Editor for Frontiers in Chemistry ISSN: 2296-2646
![]()

Member of the Editorial Board for the Organic and Medicinal Chemistry International Journal ISSN: 2474-7610
| Summaries | Pages |
|---|---|
|
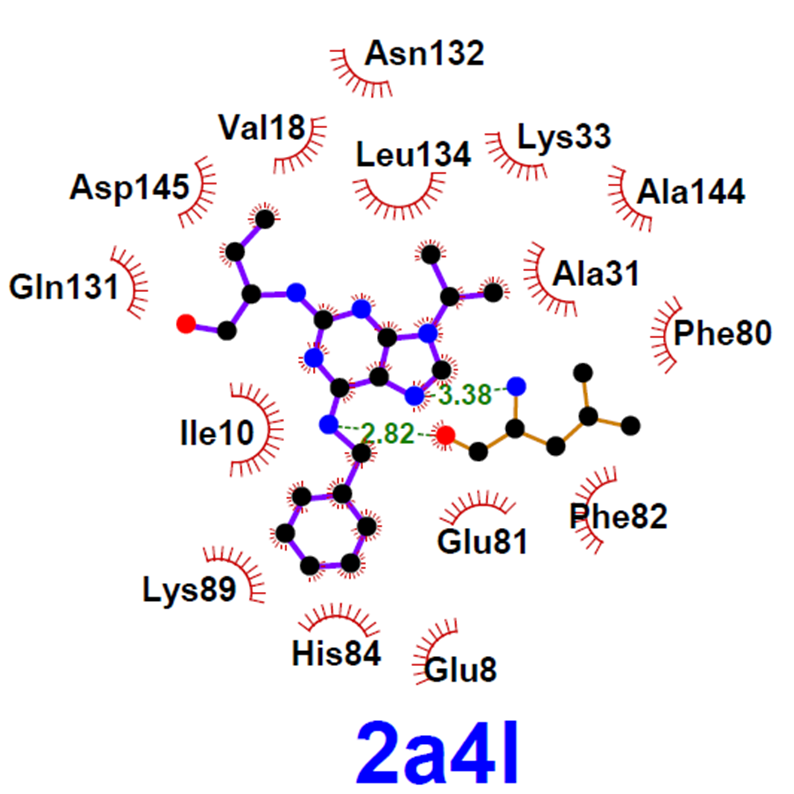
Protein-Ligand Interactions
In the study of intermolecular interactions involving protein and ligands, we expect to gain further insights into the structural basis for the specificity of small-molecule ligands against a specific protein target (de Azevedo, 2008). Analysis of protein-ligand interaction is a central problem in drug design. Knowledge of the key features responsible for the specificity of a ligand for a protein allows us to determine which physical-chemical parameters could improve the protein-ligand interaction (de Azevedo and Dias, 2008a). Furthermore, the development of a computational model to predict the binding affinity based on the atomic coordinates of a protein-ligand complex opens the possibility to apply virtual screening approaches to search small-molecule databases to identify a drug candidate (de Azevedo and Dias, 2008b, de Azevedo, 2010a; Bitencourt-Ferreira and de Azevedo, 2019a; da Silva et al., 2020). To study protein-ligand interactions, we make use of protein crystallography (Canduri and de Azevedo, 2008), nuclear magnetic resonance spectroscopy (Fadel et al., 2005), molecular docking (de Azevedo, 2010b), and molecular dynamics (de Azevedo, 2011; Bitencourt-Ferreira and de Azevedo, 2019b). References
Bitencourt-Ferreira G, de Azevedo WF Jr. Machine Learning to Predict Binding Affinity. Methods Mol Biol. 2019a; 2053: 251–273.
Bitencourt-Ferreira G, de Azevedo WF Jr. Molecular Dynamics Simulations with NAMD2. Methods Mol Biol. 2019b; 2053: 109–124.
Canduri F, de Azevedo WF. Protein crystallography in drug discovery. Curr Drug Targets. 2008; 9(12):1048–1053.
da Silva AD, Bitencourt-Ferreira G, de Azevedo WF Jr. Taba: A Tool to Analyze the Binding Affinity. J Comput Chem. 2020; 41(1): 69–73.
de Azevedo WF Jr. Protein-drug interactions. Curr Drug Targets. 2008; 9(12):1030.
de Azevedo WF Jr, Dias R. Experimental approaches to evaluate the thermodynamics of protein-drug interactions. Curr Drug Targets. 2008a; 9(12):1071–1076.
de Azevedo WF Jr, Dias R. Computational methods for calculation of ligand-binding affinity. Curr Drug Targets. 2008b; 9(12):1031–1039.
de Azevedo WF Jr. Structure-based virtual screening. Curr Drug Targets. 2010a; 11(3):261–263.
de Azevedo WF Jr. MolDock applied to structure-based virtual screening. Curr Drug Targets. 2010b; 11(3):327–334.
de Azevedo WF Jr. Molecular dynamics simulations of protein targets identified in Mycobacterium tuberculosis. Curr Med Chem. 2011; 18(9):1353–1366.
Fadel V, Bettendorff P, Herrmann T, de Azevedo WF Jr, Oliveira EB, Yamane T, Wüthrich K. Automated NMR structure determination and disulfide bond identification of the myotoxin crotamine from Crotalus durissus terrificus. Toxicon. 2005; 46(7):759–767.
Keywords: Protein; ligand; interactions; protein-ligand interactions; drug design; drug discovery; protein crystallography; nuclear magnetic resonance spectroscopy; molecular docking; molecular dynamics; simulations; docking simulations; binding affinity. |
|
|
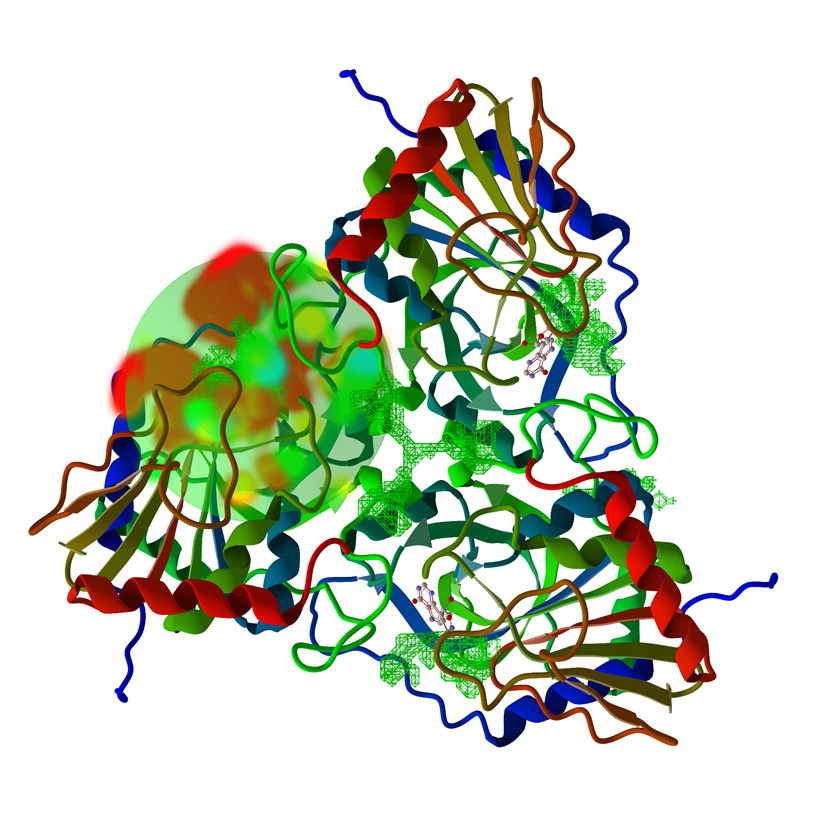
Computational Systems Biology
We have been working on the development of computational models for unraveling the molecular mechanisms underlying enzyme inhibition and protein-ligand interactions (Bitencourt-Ferreira and de Azevedo, 2019a; 2019b; 2019c; da Silva et al., 2020). We can use these computational models to predict the binding affinity of a potential inhibitor for an enzyme; such knowledge has the potential to speed up drug discovery and decrease the cost of development of new drugs (de Ávila et al., 2017; Pintro and de Azevedo, 2017). Furthermore, the availability of computational models to predict binding affinity based on the atomic coordinates of protein-ligand complexes adds flexibility to the process of drug discovery (Xavier et al., 2016; Heck et al., 2017). It allows us to computationally test different scenarios where a potential new drug may interact with a protein target. We developed the programs SAnDReS (Xavier et al., 2016) and Taba (da Silva et al., 2020) to create machine-learning models targeted to the biological system of interest. We have successfully employed SAnDReS to study coagulation factor Xa (Xavier et al., 2016), cyclin-dependent kinases (de Ávila et al., 2017; Levin et al., 2018), HIV-1 protease (Pintro and de Azevedo, 2017), estrogen receptor (Amaral et al., 2018), cannabinoid receptor 1 (Russo and de Azevedo, 2019), and 3-dehydroquinate dehydratase (de Ávila and de Azevedo, 2018). Also, we used SAnDReS to develop a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes (Bitencourt-Ferreira and de Azevedo Jr., 2018). References
Amaral MEA, Nery LR, Leite CE, de Azevedo Junior WF, Campos MM. Pre-clinical effects of metformin and aspirin on the cell lines of different breast cancer subtypes. Invest New Drugs. 2018; 36(5): 782–796.
Bitencourt-Ferreira G, de Azevedo Jr. WF. Development of a machine-learning model to predict Gibbs free energy of binding for protein-ligand complexes. Biophys Chem. 2018; 240: 63–69.
Bitencourt-Ferreira G, de Azevedo WF Jr. Machine Learning to Predict Binding Affinity. Methods Mol Biol. 2019a; 2053: 251–273.
Bitencourt-Ferreira G, de Azevedo WF Jr. Exploring the Scoring Function Space. Methods Mol Biol. 2019b; 2053: 275–281.
Bitencourt-Ferreira G, de Azevedo WF Jr. How Docking Programs Work. Methods Mol Biol. 2019c; 2053: 35–50.
da Silva AD, Bitencourt-Ferreira G, de Azevedo WF Jr. Taba: A Tool to Analyze the Binding Affinity. J Comput Chem. 2020; 41(1): 69–73.
de Ávila MB, Xavier MM, Pintro VO, de Azevedo WF. Supervised machine learning techniques to predict binding affinity. A study for cyclin-dependent kinase 2. Biochem Biophys Res Commun. 2017; 494: 305–310.
de Ávila MB, de Azevedo WF Jr. Development of machine learning models to predict inhibition of 3-dehydroquinate dehydratase. Chem Biol Drug Des. 2018; 92: 1468–1474.
Heck GS, Pintro VO, Pereira RR, de Ávila MB, Levin NMB, de Azevedo WF. Supervised Machine Learning Methods Applied to Predict Ligand-Binding Affinity. Curr Med Chem. 2017; 24(23): 2459–2470.
Levin NMB, Pintro VO, Bitencourt-Ferreira G, Mattos BB, Silvério AC, de Azevedo Jr. WF. Development of CDK-targeted scoring functions for prediction of binding affinity. Biophys Chem. 2018; 235: 1–8.
Pintro VO, Azevedo WF. Optimized Virtual Screening Workflow. Towards Target-Based Polynomial Scoring Functions for HIV-1 Protease. Comb Chem High Throughput Screen. 2017; 20(9): 820–827.
Russo S, de Azevedo WF. Advances in the Understanding of the Cannabinoid Receptor 1 - Focusing on the Inverse Agonists Interactions. Curr Med Chem. 2019; 26(10): 1908–1919.
Xavier MM, Heck GS, de Avila MB, Levin NM, Pintro VO, Carvalho NL, Azevedo WF Jr. SAnDReS a Computational Tool for Statistical Analysis of Docking Results and Development of Scoring Functions. Comb Chem High Throughput Screen. 2016; 19(10): 801–812.
Keywords: Computational systems biology; systems biology; systems approach; machine learning; protein; ligand; interactions; protein-ligand interactions; drug design; drug discovery; binding affinity. |
|
|
Molecular Simulations
Our molecular world challenges us on an everyday basis to establish the theoretical foundations to understand it. From biological molecules to the next generation of nanomaterials, the possibility of simulating them is compelling. The impact of our scientific and technological development inspires a new generation of scientists to create new computational models to simulate molecules. The success of quantum mechanics makes it clear that it is our ultimate goal to simulate molecules. On the other hand, it is evident that computer power only limits simulations of polymers. Therefore, the development and application of classical and hybrid methodologies still have a beneficial impact to assess the behavior of molecules (de Azevedo and Dias, 2008). In our research, we focus on the development of a new generation of force fields targeted to the molecular system of interest (de Azevedo, 2011). We have been working on the development of a new computational tool to study biomolecules (de Azevedo et al., 2001), nanomaterials, and the interaction between them (Bitencourt-Ferreira and de Azevedo, 2019). References
Bitencourt-Ferreira G, de Azevedo WF Jr. Molecular Dynamics Simulations with NAMD2. Methods Mol Biol. 2019; 2053: 109–124.
de Azevedo WF Jr, Canduri F, Fadel V, Teodoro LG, Hial V, Gomes RA. Molecular model for the binary complex of uropepsin and pepstatin. Biochem Biophys Res Commun. 2001; 287(1): 277–281.
de Azevedo WF Jr, Dias R. Computational methods for calculation of ligand-binding affinity. Curr Drug Targets. 2008; 9(12): 1031–1039.
de Azevedo WF Jr. Molecular dynamics simulations of protein targets identified in Mycobacterium tuberculosis. Curr Med Chem. 2011; 18(9): 1353–1366.
Keywords: Molecular simulations; computer simulations; drug design; drug discovery; molecular docking; molecular dynamics; docking simulations; binding affinity. |
|